Western blotting (‘protein blotting’ or ‘immunoblotting’) has become a powerful tool in the study of proteins.
Researchers and scientists use the western blot to identify, quantify, and determine the size of specific proteins, which is integral to research in several scientific and medical fields, particularly cancer care. Since the postdoctoral researcher W Neal Burnette coined the western blotting technique four decades ago, its popularity has grown exponentially. Today, almost all biochemistry labs around the world use the western blot.
Western blotting’s name is a play on the Southern blot (a method used to detect specific DNA sequences in DNA fragments through gel electrophoresis) and the northern blot (a method used to detect and quantify RNA, also involving gel electrophoresis). During the 1970s, Harry Towbin and his colleagues at the Friedrich Miescher Institute (Switzerland) electrophoretically separated proteins using polyacrylamide-urea gels, which they transferred onto a nitrocellulose membrane. Then, Burnette utilized the more widely used sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) to develop western blotting in 1981. The western blotting technique has evolved ever since.
As western blotting continues to shape the study of proteins, the life sciences journal BioTechniques publishes many insights into technique improvements and troubleshooting methods. Here, BioTechniques breaks down the basics of how western blotting works.
Jump to paragraph
Preparing the Sample
Researchers can measure protein for western blotting from whole tissue or tissue culture extracts. They may freeze cells and tissues with liquid nitrogen to prevent protease degradation of the proteins. However, they should avoid repeated freezing and thawing proteins, as this can weaken the quality of the protein. Alternatively, researchers may collect and lyse cells and tissues by using a homogenizer or by sonication in a lysis buffer.
Preparing Lysis Buffers and Measuring Protein Concentration
Researchers may use various detergents, buffers, and salts to enable the lysis of cells and solubilize proteins for western blotting. Using lysis buffers in the sample preparation for a western blot should improve the efficiency of protein extraction and maintain antisera recognition of the protein. Quantification and comparison with other samples in western blotting depend on the protein lysates that researchers prepare for polyacrylamide gel electrophoresis.
If using buffers, researchers should select these based on the protein of interest. They may also include protease and phosphatase inhibitors in the lysis buffer to prevent the degradation of the protein. Lots of these inhibitors come in tablet form, which means researchers can dissolve them in the lysis buffer.
Researchers must determine the protein concentrations of the samples that they will load onto a gel. They can achieve this by measuring samples at 280 nm on a spectrophotometer. They must ensure that the buffer doesn’t contain absorbing materials.
Performing Gel Electrophoresis
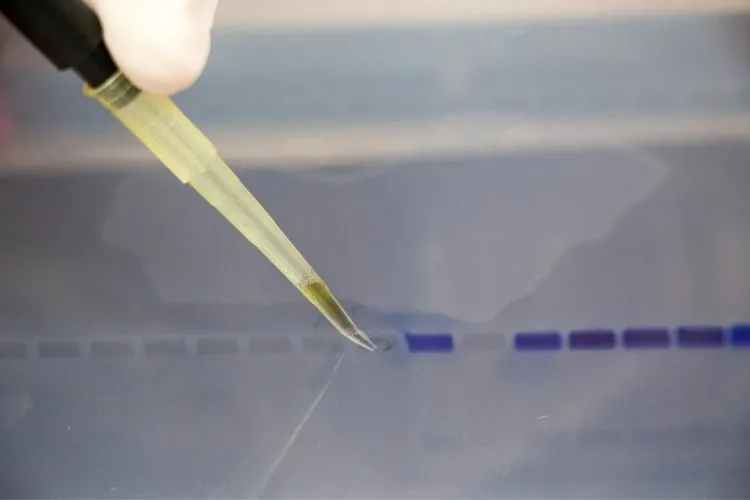
To achieve optimal separation, researchers should determine the ideal ratio of bisacrylamide and acrylamide before performing gel electrophoresis. They can then separate the proteins by molecular weight, isoelectric point, electric charge, or a combination of these.
The most common type of electrophoresis uses polyacrylamide gels and buffers that are loaded with SDS, an anionic detergent. Researchers should treat the mixture with SDS because proteins have different electrical charges. Treating the mixture with SDS denatures the proteins and gives them a negative charge. Researchers can also boil samples and apply a treatment with a reducing agent to eliminate disulfide bonds and facilitate denaturing.
By applying a voltage to the gel, researchers can make proteins migrate at different rates. The different speeds can cause the proteins to separate into bands within each lane.
Researchers may opt for a two-dimensional gel, which spreads out proteins from a sample in two dimensions. The gel separates proteins by isoelectric point in the first dimension and molecular weight in the second.
While SDS-PAGE gels unfold and denature a protein’s native structure, nondenaturing/native gels maintain protein complexes for detection after transfer. However, as complexes can’t move through polyacrylamide gels as quickly as individual denatured proteins, they might not separate cleanly or predictably.
Loading Gels
When a researcher loads samples onto a gel, they should ensure that one lane includes a molecular weight marker. Researchers should use this marker to determine the molecular weight of the target protein. Another lane should include internal control, preferably with a known molecular weight and concentration so the researcher can identify whether the primary antibody is effective.
Transferring Proteins
Once researchers have completed the electrophoresis process, they can transfer the proteins from the gel to a membrane (this membrane is the western blot). The membrane may be made of polyvinylidene difluoride, activated paper, activated nylon, or nitrocellulose, which is the most common.
Electroblotting is the most frequently used process for transferring proteins from gel to membrane because of its speed and completeness of transfer. The process involves using an electric current to pull proteins from gel to membrane. Researchers may achieve this through the immersion of a gel-membrane sandwich (wet transfer) or by securing the gel-membrane sandwich between sheets of absorbent paper that have been soaked in transfer buffer (semidry transfer).
The efficacy of protein transfer depends on the type of gel, the type of membrane, and the protein’s molecular mass. Limitations of protein transfer could include problems associated with using a transfer buffer that has a lower pH than the protein’s isoelectric point, a lower molecular weight limit of ∼10 kDa, and the use of specialized transfer buffers to facilitate the transfer of proteins that have a high isoelectric point.
Blocking Buffers and Antibodies
Researchers should prevent interactions between the membrane and the antibody they have selected to detect the target protein. They can achieve this by placing the membrane in a dilute solution of protein like nonfat dry milk or serum albumin. The blocking buffer should be appropriate for both the specific antiserum and the type of membrane. Blocking helps researchers mask potential nonspecific binding sites on the membrane. This minimizes background noise in the final product of the western blot, which removes false positives and enables clear results.
Next, researchers may incubate the membrane with the primary antibody; wash, reblock, and incubate the membrane with the secondary antibody; and then wash the membrane again. They must determine the optimal concentration of each antibody before running the samples because optimization is a prime determinant of an assay’s sensitivity. They should optimize the antibody concentration to achieve an ideal signal-to-noise ratio and may use either monoclonal or polyclonal antibodies, both of which are suitable for western blotting.
Detecting Probes
Researchers need to detect the probes that are labeled and bound to the protein of interest on the western blot. They may use chemiluminescent, colorimetric, fluorescent, or radioactive methods to achieve this. Chemiluminescent detection is the most widely used: Researchers bind the primary antibody to the protein of interest and use the secondary antibody (which is usually linked to horseradish peroxidase) to cleave a chemiluminescent agent. The reaction product produces luminescence, which is related to the protein quantity. Researchers only need a single light detector and can detect the light through photographic film or by a charged-couple device camera. These cameras are more sensitive and offer better resolution and a bigger range of exposures than film.
Once researchers have captured the exposures, they can wash the blots in a buffer and then ‘strip’ these. The stripping process involves removing bound antisera so researchers can use the blots again. Researchers can then store the blots for future reprobing. That said, repeated probing can interfere with protein antigens and result in decreased signal.
Analyzing Images
Once researchers have generated an image from a blot, they can analyze the image by densitometry to measure the relative quantity of a protein on a blot against a control or specific time point. This quantification is important if researchers need to compare samples. They should aim to achieve an exposure of the image where the bands are sharp. When comparing samples, researchers should run all samples on the same blot as there can be variation among blots.
While researchers can use commercial software programs to analyze bands on film, they can also use charged-couple device cameras, which usually have their own analysis software. Imaging software enables researchers to analyze bands, lanes, and regions automatically or manually. Researchers can then obtain relative levels of protein expression by comparing ratios of intensities of a reference band or a band of known protein concentration. They can plot relative optical density units in a graph and perform statistical analysis on samples that they have converted to optical density units.
About BioTechniques
BioTechniques is one of Future Science Group’s 34 peer-reviewed, open-access journals. The publication communicates the latest developments in life science techniques, methods, and instrumentation. Scientists, lab workers, and other industry experts from all corners of the world read and use the leading journal, which has voiced progression in scientific methodologies since the launch of its first issue in 1983. At this point, BioTechniques was the only journal to delve into scientific techniques instead of treatments.
Users can also access BioTechniques’ multimedia website, which holds a trove of webinars, videos, podcasts, interviews, articles, and eBooks. Here, experts from several scientific fields share their insights to improve collaboration and communication over developments on the life sciences scene.